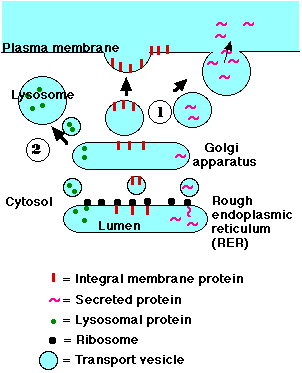
Lysosomes are roughly spherical bodies enclosed by a single membrane. They are manufactured by the Golgi apparatus (pathway 2 in the figure).
| Link to a discussion of how proteins synthesized in the endoplasmic reticulum are sent to the appropriate destinations. |
The pH within the lysosome is about pH 5, substantially less than that of the cytosol (~pH 7.2). All the enzymes in the lysosome work best at an acid pH. This reduces the risk of their digesting their own cell if they should escape from the lysosome.
| At one time, it was thought that lysosomes were responsible for killing cells scheduled to be removed from a tissue; for example, the resorption of its tail as the tadpole metamorphoses into a frog. This is incorrect. These examples of programmed cell death (PCD) or apoptosis take place by an entirely different mechanism. Link to a discussion of apoptosis. |
Materials within the cell scheduled for digestion are first deposited within lysosomes. These may be:
| Some tricks that intracellular parasites use to avoid destruction by lysosomes. |
| Link to discussion of how antigens are taken up by macrophages and B cells and degraded in their lysosomes. |
Lysosomal storage diseases are caused by the accumulation of macromolecules (proteins, polysaccharides, lipids) in the lysosomes because of a genetic failure to manufacture an enzyme needed for their breakdown. Neurons of the central nervous system are particularly susceptible to damage.
Most of these diseases are caused by the inheritance of two defective alleles of the gene encoding one of the hydrolytic enzymes.
Examples:
However, one lysosomal storage disease, I-cell disease ("inclusion-cell disease"), is caused by a failure to "tag" (by phosphorylation) all the hydrolytic enzymes that are supposed to be transported from the Golgi apparatus to the lysosomes. Lacking the mannose 6-phosphate (M6P) tag, they are secreted from the cell instead.
| Discussion of how proteins are targeted to lysosomes. |
The result: all the macromolecules incorporated in lysosomes remain undegraded forming "inclusion bodies" in the cell.
In some cells, lysosomes have a secretory function — releasing their contents by exocytosis.
Peroxisomes are about the size of lysosomes (0.5–1.5 µm) and like them are enclosed by a single membrane. They also resemble lysosomes in being filled with enzymes.
In humans, new peroxisomes are formed by the fusion of vesicles released by the endoplasmic reticulum with vesicles released by mitochondria. Once formed, peroxisomes can then increase their number by growth and division.
The enzymes and other proteins destined for peroxisomes are synthesized in the cytosol. Each contains a peroxisomal targeting signal (PTS) that binds to a receptor molecule that takes the protein into the peroxisome and then returns for another load.
Two peroxisomal targeting signals have been identified:
Some of the functions of the peroxisomes in the human liver:
Example: X-linked adrenoleukodystrophy (X-ALD). This disorder results from a failure to metabolize fatty acids properly. One result is deterioration of the myelin sheaths of neurons. The disorder occurs in young boys because the gene is X-linked. An attempt to find an effective treatment was the subject of the 1992 film Lorenzo's Oil. Since then a number of X-ALD patients have been successfully treated by gene therapy.
Example: Zellweger syndrome. This disorder results from the inheritance of two mutant genes for one of the receptors (PXR1) needed to import proteins into the peroxisome.
Peroxisomes are also called microbodies.
| Link to schematic showing lysosomes and peroxisomes in a typical animal cell. |
| Welcome&Next Search |